“批文清查三部曲”之一:上市后再评价的未来棋局
2017-03-03 20:55:01
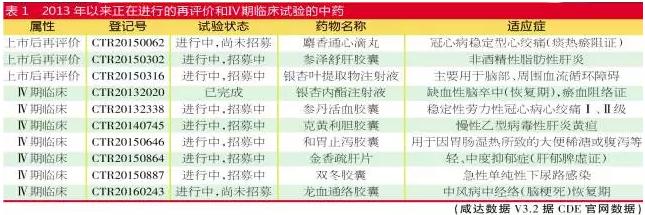 以上数据表明,2010年以后上市的独家中药口服药大多会启动上市后再评价项目。相对而言,2002年上市的中药注射剂由于生产家数多,竞争大,因此没有多大动力去履行上市再评价项目。 趋势:未来的几个重要政策导向 药品上市后再评价是1个长时间、延续的进程。虽然依照现有的药事管理制度,监管部门在药品上市前的研发、审批阶段已对该药品的风险与收益平衡机制做了评估,但出于药品的安全性等需求,药品上市后仍需要继续评价。而药品的不良反应有可能随着用量的增加逐步被暴露。另外,上市多年的“老药”也要监测其与其他药品联合使用时出现新不良反应的可能性。 美日经验:他山之石的鉴戒与启发 先来看看美国经验。美国通过药物监察项目搜集报告,建立药品不良事件报告系统数据库;评估药品商品名、标签、包装,避免处方、分发和药品管理中出现的医治毛病,以免因药品商品名与已上市的其他产品类似而产生的使用毛病。另外,美国药品评价与研究中心还建立了“药品再评价质量管理规范”,该规范包括对药品审评的进程、程序、内容、管理方式等诸多方面的规定,不单单局限于上市后药品再评价工作。 日本则将上市后药品再评价工作分为“定期再评价”和“临时再评价”两种模式。“定期再评价”是通过国内外文献调研做出是不是进行再评价的挑选,在挑选进程中,会挑选出有问题的文献,并对问题文献进行综合整理,拜托企业调查。受拜托企业提交《文献调查报告书》后,由“药事食品卫生审议会”肯定是不是进行再评价。“临时再评价”是针对紧急情况采取的评价措施。 我国政策:关联药典淘汰机制,未来纳入再评价的方向 为何新获批的中药新药需要启动上市后再评价?主要是为了让药品进入国家基本药物目录、医疗保险目录遴选,提供上市后再评价的临床研究数据将有助于厂家证明其药物的临床地位。 2016年2月14日,国务院召开常务会议,部署推动医药产业创新升级,会议肯定加大中医药投入和政策扶持,在国家基本药物目录中增加中成药品种数量。中药的政策利好,更是推动了独家中药增加再评价研究的投入。 2020年版《中国药典》近期亦开始启动编辑。2020版药典将以临床价值为导向,重点关注临床用量大、安全风险高、质量问题较多的品种,结合国家药品标准清算,建立标准淘汰机制。1是对现有国家药品标准进行清算,不符合药典收载原则的,药典将不再予以收载;2是淘汰那些没有文号、长时间没有生产、没法控制质量的标准;3是需要对药品临床价值或风险效益进行评价的,药品再评价研究数据不佳的将要面临淘汰。这亦推动企业展开再评价项目。 从安全性的角度而言,国家下1步将有可能要求多组分生化药注射剂、中药口服药和中药配方颗粒进行上市再评价研究。中药注射剂的上市再评价有可能像化药1致性评价那样设定最后期限。部份安全性和疗效存疑、临床用量较大的药品也可能进入国家再评价研究名单。 另外,国家要求改进型药品需要证明其临床有效性,而按旧化学分类获批的改进型药品基本没有做相干的实验研究。改进型药品不适用1致性评价实验,因此政策面将有可能鼓励相干药品在1定期限内展开再评价实验。 而对国外已上市使用但国内缺少且临床急需的儿童用药品种,目前儿科临床实验的履行难度较大,未来也有可能放开注册的限制或下降临床实验门坎,但须对上市后的儿童用药进行再评价,以最大程度保障儿童用药安全,并为儿童科学、公道用药提供信息支持。
以上数据表明,2010年以后上市的独家中药口服药大多会启动上市后再评价项目。相对而言,2002年上市的中药注射剂由于生产家数多,竞争大,因此没有多大动力去履行上市再评价项目。 趋势:未来的几个重要政策导向 药品上市后再评价是1个长时间、延续的进程。虽然依照现有的药事管理制度,监管部门在药品上市前的研发、审批阶段已对该药品的风险与收益平衡机制做了评估,但出于药品的安全性等需求,药品上市后仍需要继续评价。而药品的不良反应有可能随着用量的增加逐步被暴露。另外,上市多年的“老药”也要监测其与其他药品联合使用时出现新不良反应的可能性。 美日经验:他山之石的鉴戒与启发 先来看看美国经验。美国通过药物监察项目搜集报告,建立药品不良事件报告系统数据库;评估药品商品名、标签、包装,避免处方、分发和药品管理中出现的医治毛病,以免因药品商品名与已上市的其他产品类似而产生的使用毛病。另外,美国药品评价与研究中心还建立了“药品再评价质量管理规范”,该规范包括对药品审评的进程、程序、内容、管理方式等诸多方面的规定,不单单局限于上市后药品再评价工作。 日本则将上市后药品再评价工作分为“定期再评价”和“临时再评价”两种模式。“定期再评价”是通过国内外文献调研做出是不是进行再评价的挑选,在挑选进程中,会挑选出有问题的文献,并对问题文献进行综合整理,拜托企业调查。受拜托企业提交《文献调查报告书》后,由“药事食品卫生审议会”肯定是不是进行再评价。“临时再评价”是针对紧急情况采取的评价措施。 我国政策:关联药典淘汰机制,未来纳入再评价的方向 为何新获批的中药新药需要启动上市后再评价?主要是为了让药品进入国家基本药物目录、医疗保险目录遴选,提供上市后再评价的临床研究数据将有助于厂家证明其药物的临床地位。 2016年2月14日,国务院召开常务会议,部署推动医药产业创新升级,会议肯定加大中医药投入和政策扶持,在国家基本药物目录中增加中成药品种数量。中药的政策利好,更是推动了独家中药增加再评价研究的投入。 2020年版《中国药典》近期亦开始启动编辑。2020版药典将以临床价值为导向,重点关注临床用量大、安全风险高、质量问题较多的品种,结合国家药品标准清算,建立标准淘汰机制。1是对现有国家药品标准进行清算,不符合药典收载原则的,药典将不再予以收载;2是淘汰那些没有文号、长时间没有生产、没法控制质量的标准;3是需要对药品临床价值或风险效益进行评价的,药品再评价研究数据不佳的将要面临淘汰。这亦推动企业展开再评价项目。 从安全性的角度而言,国家下1步将有可能要求多组分生化药注射剂、中药口服药和中药配方颗粒进行上市再评价研究。中药注射剂的上市再评价有可能像化药1致性评价那样设定最后期限。部份安全性和疗效存疑、临床用量较大的药品也可能进入国家再评价研究名单。 另外,国家要求改进型药品需要证明其临床有效性,而按旧化学分类获批的改进型药品基本没有做相干的实验研究。改进型药品不适用1致性评价实验,因此政策面将有可能鼓励相干药品在1定期限内展开再评价实验。 而对国外已上市使用但国内缺少且临床急需的儿童用药品种,目前儿科临床实验的履行难度较大,未来也有可能放开注册的限制或下降临床实验门坎,但须对上市后的儿童用药进行再评价,以最大程度保障儿童用药安全,并为儿童科学、公道用药提供信息支持。
推荐阅读
-
 最红女星日版奶茶妹妹一缺陷被评在中国出不
最红女星日版奶茶妹妹一缺陷被评在中国出不不知道大家看过一部日本励志电影《垫底辣妹》吗?这部电影于2015年5月1日在日本上映,2016年4月14日在中国上映,...[详细]
-
 鹿晗关晓彤关系再升级两人同佩戴同款戒指
鹿晗关晓彤关系再升级两人同佩戴同款戒指鹿晗与关晓彤这对情侣自从公开恋情后经常被友扒出恩爱细节,不过近日鹿晗与关晓彤的关系疑从恋人再升级,两...[详细]
-
 明星街拍最美的刘雯樱花树下
明星街拍最美的刘雯樱花树下此页面是否是列表页或首页?未找到合适正文内容。种猪价格宝宝风寒感冒怎么办婴儿消化不良...[详细]
-
 谁能解释一下为什么石原里美顶一个油头也那
谁能解释一下为什么石原里美顶一个油头也那本文转载自OinFashion十元妹纸又出新电视剧——是一部以法医为题材的职场电视剧《UNNATURAL》又名《非自然死亡》电...[详细]
-
 女性向综艺盘点年龄段全幅化类型多样化垂直
女性向综艺盘点年龄段全幅化类型多样化垂直一线导读:女性群体成了观察的主题,而女性观众则成了锁定的目标,但在许多综艺中,女性仍然是 花瓶 角色,且...[详细]
-
 农农见老板范玮琪陈建州与陈立农合照笑容满
农农见老板范玮琪陈建州与陈立农合照笑容满陈立农范玮琪范玮琪、陈建州与陈立农四人合照 范范范瑋琪 嗨!@陈立农 农农~ 新浪娱乐讯 5月14日,范玮琪[微...[详细]
图文聚焦