一致性评价核查指导原则四连发!核查要点有哪些?
2017-01-13 10:43:30
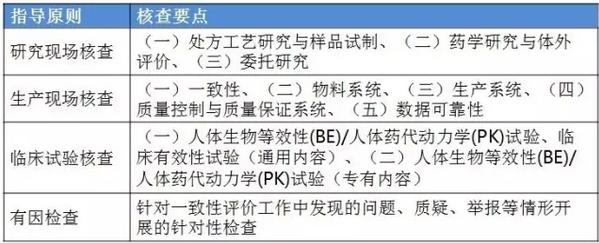 表2:4个指点原则的判定原则
表2:4个指点原则的判定原则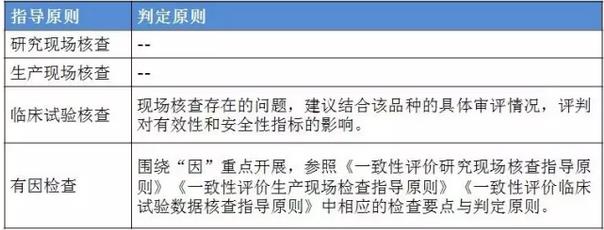 仿造药行将进入临床、研发和生产都寻求真实、合规的年代。 与“工作程序”的对应关系 这个4个原则对应2016年5月25日CFDA组织发布的《仿造药质量和疗效1致性评价工作程序》(简称“工作程序”,下同)比较以下: 研究现场核对指点原则和生产现场检查指点原则对应“工作程序”第4部份“资料的接收和受理”的研制现场核对和生产现场检查,此部份内容类似于FDA药品申报注册批准前检查。 临床实验核对指点原则和有因检查指点原则分别对应工作程序第5部份“临床实验数据核对”中临床研究数据的真实性、规范性和完全性的核对,和核对中心根据1致性评价办公室在1致性评价技术评审进程中发现的问题所展开的有因核对。 基本要求 研究现场核对指点原则和生产现场核对指点原则基本要求都包括真实性、1致性、数据可靠性和合规性。临床实验核对指点原则基本要求包括确保受试者的安全与权益得到保护,确保评价产品的1致性,确保数据的真实性、可靠性和临床实验展开的合规性。 核对程序 程序方面,进口仿造药品不管是研究现场还是生产现场都有可能要面临境外检查,核对中心将结合境外检查工作安排,在当年接收的资料当选择不低于30%的企业列入第2年的境外检查计划,组织研究现场核对。原则上在每5年内,对所有接收资料的企业的现场检查覆盖率到达100%。 有因检查的程序比较特殊。有因检查将会在1致性评价的评审进程中发现的问题,1致性评价及其药品注册相干的举报问题,药品监督管理部门或1致性评价办公室认为需进行核对的其他情形下启动。有因检查重点针对发起的缘由展开检查,可以进行必要的延伸检查,也能够不进行全面系统的检查。有因检查可采取事前通知或不告知的方式展开,也可参照飞行检查方式展开。必要时,有因检查将在现场抽取样品送1致性评价办公室指定的药品检验机构进行复核检验。 “通过”“不通过”判定原则 研究现场核对指点原则、生产现场核对指点原则和临床实验核对指点原则的判定结论都有“不通过”判定原则,这3大核对指点原则判定“不批准”共性是产生真实性;谢绝、不配合核对,致使没法继续进行现场核对都将得到“不通过”的判定。仅研究现场核对指点原则和生产现场核对指点原则有“通过”判定。临床实验核对指点原则的判定没有“通过”的判定,除“不通过”判定外只有“现场检查存在以下问题,建议结合该品种的具体审评情况,评判对有效性和安全性指标的影响”的评定结论。 有因检查比较特别,1切围绕“因”重点展开检查,但有“因”检查的“不通过”具有延续性,当有因检查的结论为“不通过”时,对应的检查结论应判定为“不通过”。 表3:4个指点原则的“通过”判定
仿造药行将进入临床、研发和生产都寻求真实、合规的年代。 与“工作程序”的对应关系 这个4个原则对应2016年5月25日CFDA组织发布的《仿造药质量和疗效1致性评价工作程序》(简称“工作程序”,下同)比较以下: 研究现场核对指点原则和生产现场检查指点原则对应“工作程序”第4部份“资料的接收和受理”的研制现场核对和生产现场检查,此部份内容类似于FDA药品申报注册批准前检查。 临床实验核对指点原则和有因检查指点原则分别对应工作程序第5部份“临床实验数据核对”中临床研究数据的真实性、规范性和完全性的核对,和核对中心根据1致性评价办公室在1致性评价技术评审进程中发现的问题所展开的有因核对。 基本要求 研究现场核对指点原则和生产现场核对指点原则基本要求都包括真实性、1致性、数据可靠性和合规性。临床实验核对指点原则基本要求包括确保受试者的安全与权益得到保护,确保评价产品的1致性,确保数据的真实性、可靠性和临床实验展开的合规性。 核对程序 程序方面,进口仿造药品不管是研究现场还是生产现场都有可能要面临境外检查,核对中心将结合境外检查工作安排,在当年接收的资料当选择不低于30%的企业列入第2年的境外检查计划,组织研究现场核对。原则上在每5年内,对所有接收资料的企业的现场检查覆盖率到达100%。 有因检查的程序比较特殊。有因检查将会在1致性评价的评审进程中发现的问题,1致性评价及其药品注册相干的举报问题,药品监督管理部门或1致性评价办公室认为需进行核对的其他情形下启动。有因检查重点针对发起的缘由展开检查,可以进行必要的延伸检查,也能够不进行全面系统的检查。有因检查可采取事前通知或不告知的方式展开,也可参照飞行检查方式展开。必要时,有因检查将在现场抽取样品送1致性评价办公室指定的药品检验机构进行复核检验。 “通过”“不通过”判定原则 研究现场核对指点原则、生产现场核对指点原则和临床实验核对指点原则的判定结论都有“不通过”判定原则,这3大核对指点原则判定“不批准”共性是产生真实性;谢绝、不配合核对,致使没法继续进行现场核对都将得到“不通过”的判定。仅研究现场核对指点原则和生产现场核对指点原则有“通过”判定。临床实验核对指点原则的判定没有“通过”的判定,除“不通过”判定外只有“现场检查存在以下问题,建议结合该品种的具体审评情况,评判对有效性和安全性指标的影响”的评定结论。 有因检查比较特别,1切围绕“因”重点展开检查,但有“因”检查的“不通过”具有延续性,当有因检查的结论为“不通过”时,对应的检查结论应判定为“不通过”。 表3:4个指点原则的“通过”判定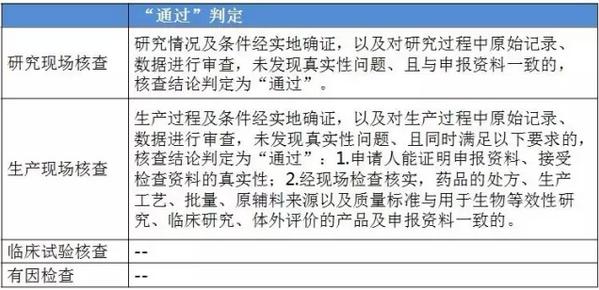 表4:4个指点原则的“不通过”判定
表4:4个指点原则的“不通过”判定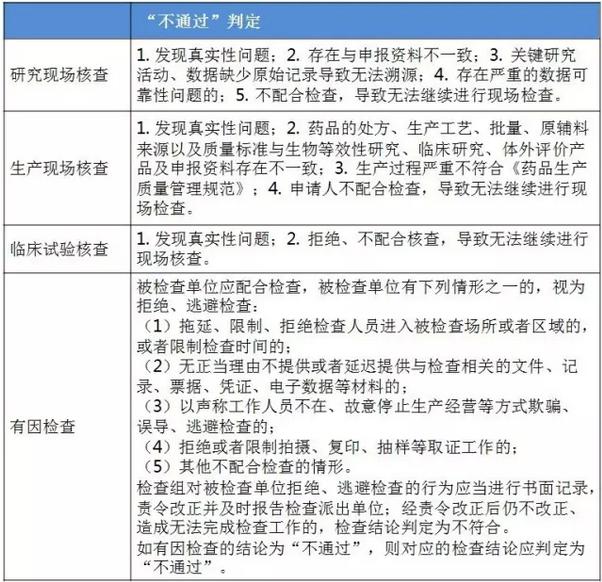 梳理<<< 1致性评价相干文件发布清单 根据《仿造药质量和疗效1致性评价工作程序》(简称“工作程序”),1致性评价工作可分为11个部份。2016年5月25日以后,这11个部份对应的文件发布汇总(见下表)可见,目前的文件发布针对从 “国家食品药品监督管理总局发布评价品种名单”(第1部份)到 “药品复核检验”(第6部份)。实际上,大部份指点原则已出台,企业应开始有针对性地根据CFDA要求准备资料。 表5:“工作程序”11个部份对应的文件发布进展汇总
梳理<<< 1致性评价相干文件发布清单 根据《仿造药质量和疗效1致性评价工作程序》(简称“工作程序”),1致性评价工作可分为11个部份。2016年5月25日以后,这11个部份对应的文件发布汇总(见下表)可见,目前的文件发布针对从 “国家食品药品监督管理总局发布评价品种名单”(第1部份)到 “药品复核检验”(第6部份)。实际上,大部份指点原则已出台,企业应开始有针对性地根据CFDA要求准备资料。 表5:“工作程序”11个部份对应的文件发布进展汇总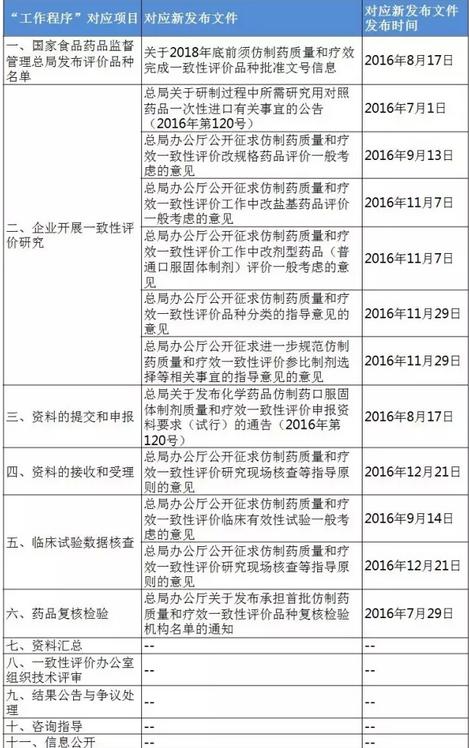
推荐阅读
-
 最红女星日版奶茶妹妹一缺陷被评在中国出不
最红女星日版奶茶妹妹一缺陷被评在中国出不不知道大家看过一部日本励志电影《垫底辣妹》吗?这部电影于2015年5月1日在日本上映,2016年4月14日在中国上映,...[详细]
-
 鹿晗关晓彤关系再升级两人同佩戴同款戒指
鹿晗关晓彤关系再升级两人同佩戴同款戒指鹿晗与关晓彤这对情侣自从公开恋情后经常被友扒出恩爱细节,不过近日鹿晗与关晓彤的关系疑从恋人再升级,两...[详细]
-
 明星街拍最美的刘雯樱花树下
明星街拍最美的刘雯樱花树下此页面是否是列表页或首页?未找到合适正文内容。种猪价格宝宝风寒感冒怎么办婴儿消化不良...[详细]
-
 谁能解释一下为什么石原里美顶一个油头也那
谁能解释一下为什么石原里美顶一个油头也那本文转载自OinFashion十元妹纸又出新电视剧——是一部以法医为题材的职场电视剧《UNNATURAL》又名《非自然死亡》电...[详细]
-
 女性向综艺盘点年龄段全幅化类型多样化垂直
女性向综艺盘点年龄段全幅化类型多样化垂直一线导读:女性群体成了观察的主题,而女性观众则成了锁定的目标,但在许多综艺中,女性仍然是 花瓶 角色,且...[详细]
-
 农农见老板范玮琪陈建州与陈立农合照笑容满
农农见老板范玮琪陈建州与陈立农合照笑容满陈立农范玮琪范玮琪、陈建州与陈立农四人合照 范范范瑋琪 嗨!@陈立农 农农~ 新浪娱乐讯 5月14日,范玮琪[微...[详细]
图文聚焦